The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line
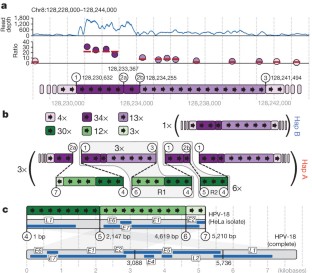
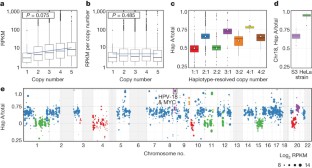
The HeLa cell line was established in 1951 from cervical cancer cells taken from a patient, Henrietta Lacks. This was the first successful attempt to immortalize human-derived cells in vitro 1 . The robust growth and unrestricted distribution of HeLa cells resulted in its broad adoption—both intentionally and through widespread cross-contamination 2 —and for the past 60 years it has served a role analogous to that of a model organism 3 . The cumulative impact of the HeLa cell line on research is demonstrated by its occurrence in more than 74,000 PubMed abstracts (approximately 0.3%). The genomic architecture of HeLa remains largely unexplored beyond its karyotype 4 , partly because like many cancers, its extensive aneuploidy renders such analyses challenging. We carried out haplotype-resolved whole-genome sequencing 5 of the HeLa CCL-2 strain, examined point- and indel-mutation variations, mapped copy-number variations and loss of heterozygosity regions, and phased variants across full chromosome arms. We also investigated variation and copy-number profiles for HeLa S3 and eight additional strains. We find that HeLa is relatively stable in terms of point variation, with few new mutations accumulating after early passaging. Haplotype resolution facilitated reconstruction of an amplified, highly rearranged region of chromosome 8q24.21 at which integration of the human papilloma virus type 18 (HPV-18) genome occurred and that is likely to be the event that initiated tumorigenesis. We combined these maps with RNA-seq 6 and ENCODE Project 7 data sets to phase the HeLa epigenome. This revealed strong, haplotype-specific activation of the proto-oncogene MYC by the integrated HPV-18 genome approximately 500 kilobases upstream, and enabled global analyses of the relationship between gene dosage and expression. These data provide an extensively phased, high-quality reference genome for past and future experiments relying on HeLa, and demonstrate the value of haplotype resolution for characterizing cancer genomes and epigenomes.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
196,21 € per year
only 3,85 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
Pan-cancer analysis of whole genomes
Article Open access 05 February 2020
3C methods in cancer research: recent advances and future prospects
Article Open access 25 April 2024

Haplotype-aware analysis of somatic copy number variations from single-cell transcriptomes
Article 26 September 2022
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
The Whole Genome Shotgun projects have been deposited in the Third Party Assembly Section of GenBank under the accessions DAAG00000000 and DAAH00000000. The versions described in this paper are versions DAAG01000000 and DAAH01000000. The sequences, variant calls, phase annotation and haplotype-specific reference sequences are available in the NIH database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap) under accession phs000642.v1.p1.
References
- Gey, G. O., Coffman, W. D. & Kubicek, M. T. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res.12, 264–265 (1952) Google Scholar
- Gartler, S. M. Apparent Hela cell contamination of human heteroploid cell lines. Nature217, 750–751 (1968) ArticleADSCASGoogle Scholar
- Skloot, R. The Immortal Life of Henrietta Lacks. (Crown Publishers, 2010) Google Scholar
- Macville, M. et al. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res.59, 141–150 (1999) CASPubMedGoogle Scholar
- Kitzman, J. O. et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nature Biotechnol.29, 59–63 (2011) ArticleCASGoogle Scholar
- Nagaraj, N. et al. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol.7, 548 (2011) ArticleGoogle Scholar
- Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature489, 57–74 (2012) ArticleADSCASGoogle Scholar
- Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science338, 222–226 (2012) ArticleADSCASGoogle Scholar
- The 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature491, 56–65 (2012) ArticleGoogle Scholar
- Exome Variant Server. http://evs.gs.washington.edu/EVS/ (NHLBI GO Exome Sequencing Project (ESP), January 2012)
- Morin, R. et al. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques45, 81–94 (2008) ArticleCASGoogle Scholar
- The Cancer Genome Project. http://www.sanger.ac.uk/genetics/CGP/ (Wellcome Trust Sanger Institute, January 2013)
- Goodwin, E. C. et al. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl Acad. Sci. USA97, 10978–10983 (2000) ArticleADSCASGoogle Scholar
- Rosty, C. et al. Clinical and biological characteristics of cervical neoplasias with FGFR3 mutation. Mol. Cancer4, 15 (2005) ArticleGoogle Scholar
- Talora, C., Sgroi, D. C., Crum, C. P. & Dotto, G. P. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev.16, 2252–2263 (2002) ArticleCASGoogle Scholar
- White, E. A. et al. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol.86, 13174–13186 (2012) ArticleCASGoogle Scholar
- Corver, W. E. et al. Genome-wide allelic state analysis on flow-sorted tumor fractions provides an accurate measure of chromosomal aberrations. Cancer Res.68, 10333–10340 (2008) ArticleCASGoogle Scholar
- Wingo, S. N. et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS ONE4, e5137 (2009) ArticleADSGoogle Scholar
- Wistuba, I. I. et al. Deletions of chromosome 3p are frequent and early events in the pathogenesis of uterine cervical carcinoma. Cancer Res.57, 3154–3158 (1997) CASPubMedGoogle Scholar
- Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell149, 994–1007 (2012) ArticleCASGoogle Scholar
- Fan, H. C., Wang, J., Potanina, A. & Quake, S. R. Whole-genome molecular haplotyping of single cells. Nature Biotechnol.29, 51–57 (2011) ArticleCASGoogle Scholar
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature489, 519–525 (2012); corrigendum. 491, 288 (2012)
- Puck, T. T. & Marcus, P. I. A rapid method for viable cell titration and clone production with HeLa cells in tissue culture: the use of X-irradiated cells to supply conditioning factors. Proc. Natl Acad. Sci. USA41, 432–437 (1955) ArticleADSCASGoogle Scholar
- Nelson-Rees, W. A., Daniels, D. W. & Flandermeyer, R. R. Cross-contamination of cells in culture. Science212, 446–452 (1981) ArticleADSCASGoogle Scholar
- Wentzensen, N., Vinokurova, S. & von Knebel Doeberitz, M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res.64, 3878–3884 (2004) ArticleCASGoogle Scholar
- Lazo, P. A., DiPaolo, J. A. & Popescu, N. C. Amplification of the integrated viral transforming genes of human papillomavirus 18 and its 5′-flanking cellular sequence located near the myc protooncogene in HeLa cells. Cancer Res.49, 4305–4310 (1989) CASPubMedGoogle Scholar
- Bouallaga, I., Massicard, S., Yaniv, M. & Thierry, F. An enhanceosome containing the Jun B/Fra-2 heterodimer and the HMG-I(Y) architectural protein controls HPV 18 transcription. EMBO Rep.1, 422–427 (2000) ArticleCASGoogle Scholar
- Li, G. et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell148, 84–98 (2012) ArticleCASGoogle Scholar
- Peter, M. et al. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene25, 5985–5993 (2006) ArticleCASGoogle Scholar
- Ahmadiyeh, N. et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl Acad. Sci. USA107, 9742–9746 (2010) ArticleADSCASGoogle Scholar
- Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics25, 1754–1760 (2009) ArticleCASGoogle Scholar
- McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res.20, 1297–1303 (2010) ArticleCASGoogle Scholar
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics27, 2987–2993 (2011) ArticleCASGoogle Scholar
- Gymrek, M., Golan, D., Rosset, S. & Erlich, Y. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res.22, 1154–1162 (2012) ArticleCASGoogle Scholar
- Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols4, 44–57 (2009) ArticleCASGoogle Scholar
- Hach, F. et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nature Methods7, 576–577 (2010) ArticleCASGoogle Scholar
- Sudmant, P. H. et al. Diversity of human copy number variation and multicopy genes. Science330, 641–646 (2010) ArticleADSCASGoogle Scholar
- Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA108, 1513–1518 (2011) ArticleADSCASGoogle Scholar
- Talkowski, M. E. et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am. J. Hum. Genet.88, 469–481 (2011) ArticleCASGoogle Scholar
- Adey, A. et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome. Biol.11, R119 (2010) ArticleCASGoogle Scholar
- Duitama, J. et al. Fosmid-based whole genome haplotyping of a HapMap trio child: evaluation of single individual haplotyping techniques. Nucleic Acids Res.40, 2041–2053 (2012) ArticleCASGoogle Scholar
- Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics25, 1105–1111 (2009) ArticleCASGoogle Scholar
- Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics27, 2325–2329 (2011) ArticleCASGoogle Scholar
Acknowledgements
The genome sequence described in this paper was derived from a HeLa cell line. Henrietta Lacks, and the HeLa cell line that was established from her tumour cells in 1951, have made significant contributions to scientific progress and advances in human health. We are grateful to Henrietta Lacks, now deceased, and to her surviving family members for their contributions to biomedical research. We also thank M. Kircher, M. Snyder, A. Kumar and R. Patwardhan as well as other members of the Shendure laboratory for advice and suggestions. We thank the Stamatoyannopoulos and Malik laboratories for cell aliquots. Our work was supported by a gift from the Washington Research Foundation; grant HG006283 from the National Genome Research Institute (NHGRI, to J.S.); grant CA160080 from the National Cancer Institute (to J.S.); a graduate research fellowship DGE-0718124 from the National Science Foundation (to A.A. and J.K.); grant T32HG000035 from the NHGRI (to J.N.B.); and grant AG039173 from the National Institute of Aging (to J.B.H.). J.S. is the Lowell Milken Prostate Cancer Foundation Young Investigator. J.S. is a member of the scientific advisory board or serves as a consultant for Ariosa Diagnostics, Stratos Genomics, Good Start Genetics, and Adaptive Biotechnologies.
Author information
- Andrew Adey, Joshua N. Burton and Jacob O. Kitzman: These authors contributed equally to this work.
Authors and Affiliations
- Department of Genome Sciences, University of Washington, Seattle, 98115, Washington, USA Andrew Adey, Joshua N. Burton, Jacob O. Kitzman, Joseph B. Hiatt, Alexandra P. Lewis, Beth K. Martin, Ruolan Qiu, Choli Lee & Jay Shendure
- Andrew Adey
